Gene Mutations in Inherited Retinal Diseases
Written by Sayantan Mitra
Published 26th April 2024 / Last updated 26th April 2026
In a new study from LVPEI, Drs. Chitra Kannabiran, Deepika C. Parameswarappa, Deepak K. Bagga, and others analyzed various mutations in the RPE65 gene that can manifest as inherited retinal diseases.
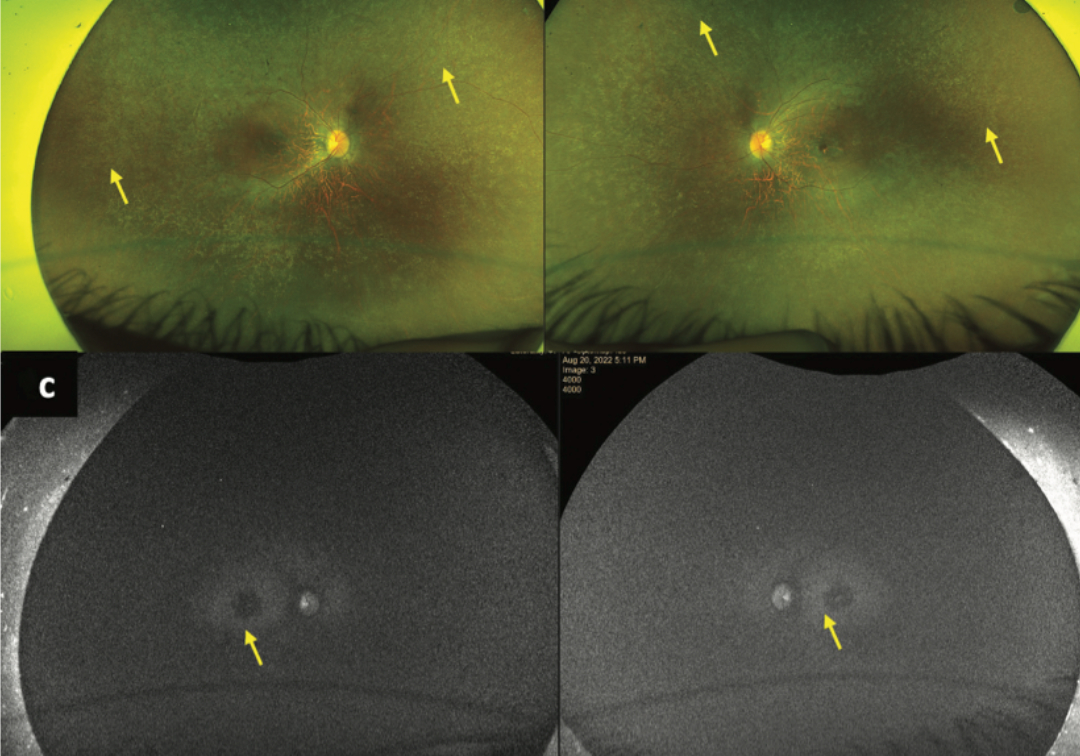
When light lands on the retina, specific proteins on photoreceptor cells (rods) transform in shape, triggering the brain’s visual processing. These proteins are regularly recycled by a set of enzymes produced and maintained by the retinal pigment epithelium (RPE), a single cell casing around the retina. In a set of rare, inherited retinal diseases (IRDs) this system is disrupted leading to severe vision loss and blindness. IRDs, triggered by genetic mutations, often manifest during infancy or early childhood. Leber congenital amaurosis (LCA), early-onset severe retinal dystrophy (EOSRD), and retinitis pigmentosa (RP) are some of the common IRDs found in Indian children.
The RPE65 gene is expressed at high levels in the retinal pigment epithelium. RPE65 encodes a protein necessary for retinol (vitamin A) cycling, a biochemical process that converts light perceived by photoreceptors into electrical signals for the brain. Mutations in RPE65 disrupt the retinol regeneration process, causing photoreceptors to degenerate and leading to IRDs such as LCA, EOSRD, and RP. Studies estimate that globally, 1-20% of LCA and 1-4% of RP is due to RPE65 mutation. But such studies rarely include Indians, which means the genetics of IRDs among Indians is unclear. Populations in southern India are also vulnerable to IRDs because of the prevalence of consanguinity, which is associated with various genetic ocular disorders including retinal dystrophies. Identifying pathogenic mutations of RPE65 can provide a better understanding of the underlying genetic factors in IRDs among Indians.
In a new study published in the journal Ophthalmic Genetics, Drs. Chitra Kannabiran, Deepika Parameswarappa, Deepak Bagga, and others report on the frequency and types of RPE65 mutations and the clinical features of patients with LCA, EOSRD, and RP. The study screened 220 patients with IRDs (150 ‘probands’—the first person studied in a family—for direct sequencing of RPE65, and 70 unrelated patients with next generation sequencing on a panel of over 260 genes associated with IRDs) and 150 healthy controls for mutations. Apart from RPE65, none of the other 260+ genes had any pathogenic mutations. The RPE65 gene was also sequenced to identify various mutations and their associated phenotypic manifestations in LCA, EOSRD, and RP.
The study included the genetic and clinical analysis of 8 probands (3.6%) with RPE65 mutations that led to IRDs. Of them, 7 patients had LCA onset before their first birthday. One patient developed EOSRD in their second year. Five of the eight (62.5%) patients had a history of parental consanguinity. The mutations in the seven patients with LCA led to a premature termination of mRNA synthesis; without a full-sized mRNA of correct sequence, there can be no fully formed protein. One patient had a missense mutation, which results in a different, and often dysfunctional, protein. The study highlights that RPE65 mutations, while rare, manifest as severe IRD, at a very young age. If identified early, and when the child still has healthy photoreceptor and RPE cells, there is the promise of gene therapy.
‘The study sheds light on the frequency of mutations in this gene in a series of over 200 cases with various forms of early-onset retinal dystrophy in our population, clinical features common to the patients with RPE65 mutations and challenges involved in testing functional vision in the patients,’ concludes Dr. Chitra Kannabiran, a scientist at LVPEI and the corresponding author of this paper. ‘These observations may aid in assessment of the utility and effective window period for treating such patients with gene therapy.’
Citation
Parameswarappa, D. C., Bagga, D. K., Upadhyaya, A., Balasubramanian, J., Pochaboina, V., Muthineni, V., Jalali, S., & Kannabiran, C. (2024). RPE65 mutations in Leber congenital amaurosis, early-onset severe retinal dystrophy, and retinitis pigmentosa from a tertiary eye care center in India. Ophthalmic Genetics, 1–10. Advance online publication. https://doi.org/10.1080/13816810.2024.2309559